상위 질문
타임라인
채팅
관점
탄소-수소 결합 활성화
위키백과, 무료 백과사전

Remove ads
유기화학 및 유기금속화학에서 탄소-수소 결합 활성화(영어: Carbon–hydrogen bond activation, C−H 활성화(영어: C−H activation))는 탄소-수소 결합이 분리되고 C−X 결합 (X ≠ H는 일반적으로 탄소, 산소 또는 질소와 같은 주족 원소)으로 대체되는 유기 반응의 한 유형이다. 일부 저자들은 C–H 활성화라는 용어를 "비활성"으로 간주되는 C–H 결합이 전이 금속 중심 M과 상호 작용하여 결합이 절단되고 M–C 결합을 가진 유기금속 종이 생성되는 반응으로 더 제한한다. 이 단계(때로는 C−H 활성화 단계로 알려짐)에서 생성된 유기금속 중간체는 다른 시약과 후속 반응을 거칠 수 있으며, 이는 그 자리에서 (종종 전이 금속이 촉매 양으로 사용되도록 허용) 또는 별도의 단계에서 기능화된 생성물을 생산한다.[1]

대체 용어인 C−H 기능화(영어: C−H functionalization)는 반응 과정에 관계없이 (또는 이에 대한 불가지론적 태도로) 비교적 비활성인 C−H 결합을 C−X 결합으로 변환하는 모든 반응을 설명하는 데 사용된다. 특히, 이 정의는 분리된 C–H 결합이 처음에 전이 금속과 상호 작용하거나 반응 메커니즘에 유기금속 중간체가 존재할 필요가 없다.[2] 유기금속 종과 달리, 이 확장된 유형의 C-H 활성화는 산업 및 자연에서 널리 사용된다. 이 광범위한 정의는 위에서 언급한 C–H 활성화의 제한된 정의에 해당하는 모든 반응을 포함한다. 그러나 산소 반발 메커니즘을 통해 진행되는 철 촉매 알케인 C–H 수산화 반응(예: 사이토크롬 P450 효소 및 그 합성 유사체)도 포함하며, 이 메커니즘에는 유기금속 종이 관여하지 않는 것으로 여겨진다. 다른 경우에는 유기금속 종이 간접적으로 관여한다. 예를 들어, Rh(II)-촉매 C–H 삽입 과정에서 친전자성 금속 카벤 종이 생성되고 탄화수소 C–H 결합이 금속과의 직접적인 상호 작용 없이 카벤 탄소에 삽입되는 경우에 발생한다. 금속에 의한 직접적인 C–H 결합 분해를 포함하지 않는 다른 메커니즘 가능성으로는 (i) 친전자성 방향족 치환 메커니즘에 의한 아릴금속 종 생성(친전자성 Pd, Pt, Au, Hg 종에 흔함), (ii) O- 또는 N-중심 라디칼에 의한 수소 원자 추상화를 통한 C–H 결합 분해(이후 유기금속 중간체를 형성하거나 형성하지 않고 더 반응하고 기능화될 수 있음, 예: 카라시-소노프스키 반응), (iii) 친전자성 금속과의 π-착물 초기 형성에 의해 보조되는 π-시스템의 α-위치에서 C–H 탈양성자화로 친핵성 유기금속 종 생성(예: 사이클로펜타디에닐아이언 착물에 의해)이 있다.
종종 저자들이 C–H 기능화와 C−H 활성화를 구분할 때, 후자를 좁은 의미로 제한한다. 그러나 C–H 결합과 금속 사이의 상호 작용이 결합이 분해되기 전에 있었는지 여부를 확실히 입증하는 것은 어려울 수 있다. 이 글은 일반적으로 C–H 기능화 반응을 다루지만, 특히 C–H 활성화에 중점을 둔다.
Remove ads
분류
금속 중심에 의한 C-H 활성화 메커니즘은 세 가지 일반적인 범주로 분류할 수 있다:
- (i) 산화성 첨가: 저원자가 금속 중심이 탄소-수소 결합에 삽입되어 결합을 분해하고 금속을 산화시킨다:
- LnM + RH → LnM(R)(H)
- (ii) 친전자성 활성화: 친전자성 금속이 탄화수소를 공격하여 양성자를 치환시킨다:
- LnM+ + RH → LnMR + H+
- 이 범주 중 특히 흔한 변형인 협동 금속화 탈양성자화는 배위된 내부 염기(종종 카르복실레이트, 예를 들어 아세테이트 또는 피발레이트)가 치환된 양성자를 분자 내에서 동시에 수용하는 것을 포함한다.
- (iii) 시그마 결합 복분해: "4중심" 전이 상태를 통해 진행되며, 이 상태에서는 결합이 한 단계에서 끊어지고 형성된다:
- LnMX + RH → LnMR + XH
Remove ads
역사적 개요
요약
관점
최초의 C–H 활성화 반응은 종종 1902년에 오토 딤로트가 벤젠이 아세트산 수은(II)과 반응한다고 보고한 것에 기인한다 (참조: 유기수은 화합물). 많은 친전자성 금속 중심이 이 프리델-크래프츠와 유사한 반응을 겪는다. 조지프 채트는 Ru(0) 착물에 의한 나프탈렌의 C-H 결합 첨가를 관찰했다.[3]
킬레이트-보조 C-H 활성화는 널리 퍼져 있다. 무라하시 슌스케는 (E)-N,1-다이페닐메탄이민으로부터 2-페닐아이소인돌린-1-온의 코발트-촉매 킬레이트-보조 C-H 기능화를 보고했다.[4]

1969년, 알렉산더 E. 실로프는 테트라클로로백금산 칼륨이 메테인과 중수 사이의 동위 원소 혼합을 유도한다고 보고했다. 이 경로는 메테인이 Pt(II)에 결합하는 것을 포함한다고 제안되었다. 1972년, 실로프 연구팀은 테트라클로로백금산 칼륨, 촉매 헥사클로로백금산 칼륨, 메테인 및 물을 화학량론적 양으로 포함하는 유사한 반응에서 메탄올과 염화 메틸을 생산할 수 있었다. 실로프가 냉전 시대에 소련에서 연구하고 발표했기 때문에 그의 연구는 서방 과학자들에게 대부분 무시되었다. 이른바 실로프 시스템은 오늘날 알케인 기능화를 위한 몇 안 되는 진정한 촉매 시스템 중 하나이다.[1][5]
어떤 경우에는 C-H 활성화의 발견이 교차 짝지음과 함께 이루어졌다. 1969년에[6] 후지와라 유조는 Pd(OAc)2와 Cu(OAc)2를 사용하여 벤젠과 스타이렌으로부터 (E)-1,2-다이페닐에텐을 합성했다고 보고했으며, 이는 교차 짝지음과 매우 유사한 절차였다. 산화적 첨가 범주에서 M. L. H. 그린은 1970년에 Cp2WH2 착물로서 텅스텐이 벤젠 C–H 결합에 광화학적으로 삽입되는 것을 보고했으며[7] 조지 M. 화이트사이드는 1979년에 최초로 분자 내 지방족 C–H 활성화를 수행했다.[8]

다음 혁신은 1982년에 두 연구팀에 의해 독립적으로 보고되었다. 로버트 G. 버그만은 산화적 첨가를 통해 비활성화되고 완전히 포화된 탄화수소의 최초 전이 금속 매개 분자간 C–H 활성화를 보고했다. 광화학적 접근법을 사용하여 Cp*Ir(PMe3)H2(여기서 Cp*는 펜타메틸사이클로펜타다이에닐 리간드)의 광분해는 배위적으로 불포화된 종 Cp*Ir(PMe3)를 생성했으며, 이는 사이클로헥세인 및 네오펜테인과 산화적 첨가를 통해 반응하여 해당 하이드리도알킬 착물 Cp*Ir(PMe3)HR을 형성했다(여기서 R은 각각 사이클로헥실 및 네오펜틸이다).[9] W.A.G. 그레이엄은 동일한 탄화수소가 조사 시 Cp*Ir(CO)2와 반응하여 관련 알킬하이드리도 착물 Cp*Ir(CO)HR을 생성한다는 것을 발견했다(여기서 R은 각각 사이클로헥실 및 네오펜틸이다).[10] 후자의 예에서 반응은 Cp*Ir(CO)2의 조사를 통해 형성된 16-전자 이리듐(I) 중간체인 Cp*Ir(CO)에 대한 알케인의 산화적 첨가를 통해 진행되는 것으로 추정된다.
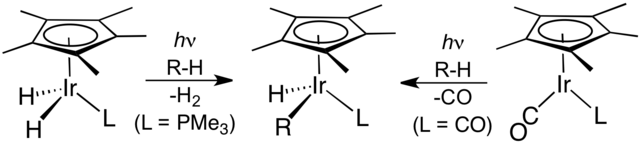
버그만 외 (왼쪽) 및 그레이엄 외에 의한 C–H 활성화.

알케인 C–H 결합의 선택적 활성화 및 기능화는 펜타메틸사이클로펜타다이에닐, 금속 나이트로실, 알릴 및 네오펜틸 리간드를 갖춘 텅스텐 착물인 Cp*W(NO)(η3-알릴)(CH2CMe3)을 사용하여 보고되었다.[11]

레그진스 외, J. Am. Chem. Soc. 2007; 129, 5372–3에 나타난 펜테인 C–H 활성화.

이 시스템과 관련된 한 예에서, 알케인인 펜테인은 선택적으로 할로겐화 탄화수소인 1-아이오도펜테인으로 전환된다. 이 변환은 실온에서 펜테인 내의 Cp*W(NO)(η3-알릴)(CH2CMe3)의 열분해를 통해 이루어졌으며, 이는 유사 1차 과정을 통해 네오펜테인의 제거를 초래하여 16-전자 중간체를 생성하는데, 이는 검출할 수 없지만 전자적으로나 입체적으로 불포화된 η2-뷰타다이엔 리간드에 의해 배위된다. 이후 펜테인 용매 분자의 분자간 활성화는 n-펜틸 리간드를 갖는 18-전자 착물을 생성한다. 별도의 단계에서, -60 °C에서 아이오딘과의 반응은 착물에서 1-아이오도펜테인을 방출한다.
Remove ads
반응 메커니즘 이해
화학 반응을 개선하는 한 가지 접근 방식은 근본적인 반응 과정을 이해하는 것이다. 시간 분해 분광 기술은 화학 반응의 동역학을 추적하는 데 사용될 수 있다. 이 기술은 과정을 시작하기 위한 트리거를 필요로 하는데, 대부분의 경우 화합물을 조명하는 것이다. 전이 금속 착물과 알케인의 광개시 반응은 강한 C-H 결합의 절단을 이해하기 위한 강력한 모델 시스템 역할을 한다.[9][10]

이러한 시스템에서 샘플은 UV 광에 노출되어 금속 중심을 여기시키고 리간드 해리를 유도한다. 이 해리는 빈 배위 위치를 가진 고도로 반응성이 있는 전자 결핍 16전자 중간체를 생성한다. 이 종은 알케인 분자와 결합하여 σ-착물을 형성한다(C-H 결합의 배위). 세 번째 단계에서 금속 원자는 C-H 결합에 삽입되어 이를 분해하고 알킬(또는 아릴) 금속 수소화물을 생성한다.
중간체와 그 동역학은 시간 분해 분광 기술 (예: TR-IR, TR-XAS, TR-RIXS)을 사용하여 관찰할 수 있다. 시간 분해 적외선 분광법 (TR-IR)은 이러한 중간체를 관찰하기에 다소 편리한 방법이다. 그러나 이는 IR-활성 리간드를 가진 착물에만 한정되며, 기본 진동 냉각으로 인해 펨토초 시간 규모에서의 정확한 할당에 취약하다. 서로 다른 착물에 대한 반응성 차이의 질문에 답하기 위해, 이들의 전자 구조를 조사해야 한다. 이는 X선 흡수 분광법 (XAS) 또는 공명 비탄성 X선 산란 (RIXS)을 통해 달성될 수 있다. 이러한 방법들은 궤도 분해능으로 C-H 활성화 단계를 추적하고 C-H 결합 분해에 책임이 있는 상호 작용에 대한 자세한 통찰력을 제공하는 데 사용되었다.[12][13]
지롤라미는 2023년에 금속 중심에 결합된 메테인의 구조를 완전히 특성화했다. 중수소화 동위 원소체를 포함하는 평형 동위 원소 섭동(IPE) 연구는 메테인이 단일 M···H-C 다리를 통해 금속 중심에 결합한다는 것을 보여주었다. 1JCH 결합 상수의 변화는 메테인 리간드의 구조가 자유 분자에 비해 상당히 교란되었음을 분명히 나타낸다.[14]
방향성 C-H 활성화
방향성, 킬레이트-보조 또는 "유도된" C-H 활성화는 위치 선택성과 입체 선택성에 영향을 미치는 지향기를 포함한다.[15] 이는 유기 합성에서 가장 유용한 C-H 활성화 방식이다. N,N-다이메틸벤질아민은 많은 전이 금속에 의해 쉽게 사이클로금속화된다.[16] 무라이 반응에서 약하게 배위하는 지향기들이 사용되는 준실용적인 구현들이 있다.[17]

2-페닐피리딘의 Pd-촉매 C-H 활성화 반응 메커니즘은 금속고리 중간체를 포함한다. 이 중간체는 산화되어 PdIV 종을 형성한 후 환원성 제거를 통해 C-O 결합을 형성하고 생성물을 방출한다.[18]

보릴화
C-H 결합을 C-B 결합으로 변환하는 보릴화는 합성(즉, 교차-짝지음 반응)에서의 유용성 때문에 철저히 연구되어 왔다. 존 F. 하트위그는 로듐 착물에 의해 촉매되는 고도로 위치선택적인 아렌 및 알케인 보릴화를 보고했다. 알케인의 경우, 독점적인 말단 기능화가 관찰되었다.[19]

나중에 루테늄 촉매는 더 높은 활성도와 작용기 호환성을 가지고 있음이 발견되었다.[20]

높은 호환성으로 C-H 결합을 활성화하는 이리듐 기반 촉매를 포함한 다른 보릴화 촉매도 개발되었다.[21][22][23]
자세한 내용은 보릴화를 참조.
Remove ads
천연가스
화학자들이 메테인의 선택적 C-H 활성화를 위한 상업적 공정을 개발하는 데 실패했지만, 이러한 반응은 역 메테인 생성의 기초가 된다. 이 니켈-촉매 과정에서 메테인은 조효소 M, CH
3SCH
2CH
2SO−
3의 메틸 치환체로 전환된다.[24]
자연적으로 발생하는 메테인은 풍부하고 저렴함에도 불구하고 화학 원료로 사용되지 않는다. 현재 기술은 수증기 개질을 통해 메테인을 엄청나게 사용하여 합성가스, 즉 일산화 탄소와 수소의 혼합물을 생산한다. 이 합성가스는 Fischer-Tropsch 반응에서 더 긴 탄소 사슬 제품이나 가장 중요한 산업 화학 원료 중 하나인 메탄올을 만드는 데 사용된다.[25][26] 이러한 탄화수소를 전환하는 흥미로운 방법은 C-H 활성화를 포함한다. 예를 들어 로이 A. 페리아나는 Pt, Pd, Au, Hg과 같은 후기 전이 금속을 포함하는 착물이 H2SO4에서 메테인 (CH4)과 반응하여 메틸 바이설페이트를 생성한다고 보고했다.[27][28] 그러나 이 과정은 상업적으로 구현되지 않았다.

Remove ads
비대칭 C–H 기능화

로듐 촉매 카벤 삽입 반응은 키랄 카르복실레이트 리간드, 특히 프롤린 유도체 DOSP 리간드를 사용하여 거울상 선택적으로 이루어졌다.[30]
리토스페르민산의 전합성은 고도로 기능화된 시스템에 대한 유도된 C-H 기능화 후기 단계를 활용한다. 카이랄 비라세미 이민인 지향기는 분자내 알킬화를 수행할 수 있으며, 이는 로듐 촉매 이민의 다이하이드로벤조퓨란으로의 전환을 가능하게 한다.[31]

거울상 선택적 C-H 실릴화는 실리콘 또는 인근 탄소 중심에 잘 정의된 입체 화학을 도입하는 유용한 전략으로 부상했다.[32][33]
천연물 및 생체 활성 분자 합성 응용
칼로트릭신 A와 B의 전합성은 C-H 활성화를 통한 분자내 Pd-촉매 교차 짝지음 반응을 특징으로 하며, 이는 유도된 C-H 활성화의 한 예이다. 교차 짝지음은 아릴 C-I와 C-H 결합 사이에서 발생하여 C-C 결합을 형성한다.[34] 메시칼린 유사체의 합성은 C-H 활성화를 통한 아릴 이민의 로듐-촉매 거울상 선택적 환상화를 활용한다.[35]

알켄 이성질화
유용한 전이 금속 C-H 결합 활성화의 한 유형은 알켄 이성질화이다. 최소한 두 가지 메커니즘이 알려져 있다. 알켄-금속 수소화물의 경우, 이성질화는 전이 삽입에 이어 베타-수소화물 제거를 통해 진행될 수 있다. 이 과정은 사슬 이동의 기초이다. 알켄 이성질화의 또 다른 메커니즘은 알켄 착물이 알릴-수소화물 착물로 전환되는 것이다.[36]
같이 보기
- 탄소-탄소 결합 활성화
- 메테인의 산화적 짝지음
- 교차 탈수소화 짝지음(CDC 반응)
- 실로프 시스템
- 메타 선택적 C-H 기능화
오래된 리뷰
- 2004년 이전
- Arndtsen, B. A.; Bergman, R. G.; Mobley, T. A.; Peterson, T. H. (1995). 《Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution.》. 《Accounts of Chemical Research》 28. 154–162쪽. doi:10.1021/ar00051a009.
- Crabtree, R. H. (2001). 《Alkane C–H activation and functionalization with homogeneous transition metal catalysts: a century of progress – a new millennium in prospect》. 《J. Chem. Soc., Dalton Trans.》 17. 2437–2450쪽. doi:10.1039/B103147N.
- 2004-7
- Crabtree, R. H. (2004). 《Organometallic alkane CH activation》. 《J. Organomet. Chem.》 689. 4083–4091쪽. doi:10.1016/j.jorganchem.2004.07.034. S2CID 95482372.
- Organometallic C–H Bond Activation: An Introduction Alan S. Goldman and Karen I. Goldberg ACS Symposium Series 885, Activation and Functionalization of C–H Bonds, 2004, 1–43
- Periana, R. A.; Bhalla, G.; Tenn, W. J.; III; Young, K. J. H.; Liu, X. Y.; Mironov, O.; Jones, C.; Ziatdinov, V. R. (2004). 《Perspectives on some challenges and approaches for developing the next generation of selective, low temperature, oxidation catalysts for alkane hydroxylation based on the C–H activation reaction》. 《Journal of Molecular Catalysis A: Chemical》 220. 7–25쪽. doi:10.1016/j.molcata.2004.05.036.
- Lersch, M.Tilset (2005). 《Mechanistic Aspects of C−H Activation by Pt Complexes》. 《Chem. Rev.》 105. 2471–2526쪽. doi:10.1021/cr030710y. PMID 15941220., Vedernikov, A. N. (2007). 《Recent Advances in the Platinum-mediated CH Bond Functionalization》. 《Curr. Org. Chem.》 11. 1401–1416쪽. doi:10.2174/138527207782418708.
- 2008-2011
- Davies, H. M. L.; Manning, J. R. (2008). 《Catalytic C–H functionalization by metalcarbenoid and nitrenoid insertion》. 《Nature》 451. 417–424쪽. Bibcode:2008Natur.451..417D. doi:10.1038/nature06485. PMC 3033428. PMID 18216847.
- Boutadla, Y.; Davies, D. L.; Macgregor, S. A.; Poblador-Bahamonde, A. I. (2009). 《Mechanisms of C–H bond activation: rich synergy between computation and experiment》. 《Dalton Trans》 2009. 5820–5831쪽. doi:10.1039/B904967C. PMID 19623381.
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. (2011). 《Towards Mild Metal-Catalyzed C–H Bond Activation》. 《Chem. Soc. Rev.》 40. 4740–4761쪽. doi:10.1039/C1CS15083A. PMID 21666903.
- Shulpin, G. B. (2010). 《Selectivity enhancement in functionalization of C–H bonds: A review》. 《Org. Biomol. Chem.》 8. 4217–4228쪽. doi:10.1039/c004223d. PMID 20593075.
- Lyons, T. W.; Sanford, M. S. (2010). 《Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions》. 《Chem. Rev.》 110. 1147–1169쪽. doi:10.1021/cr900184e. PMC 2836499. PMID 20078038.*Balcells, D.; Clot, E.; Eisenstein, O. (2010). 《C–H Bond Activation in Transition Metal Species from a Computational Perspective》. 《Chem. Rev.》 110. 749–823쪽. doi:10.1021/cr900315k. PMID 20067255.
- 2012-2015
- Hashiguchi, B. G.; Bischof, S. M.; Konnick, M. M.; Periana, R. A. (2012). 《Designing Catalysts for Functionalization of Unactivated C–H Bonds Based on the CH Activation Reaction》. 《Acc. Chem. Res.》 45. 885–898쪽. doi:10.1021/ar200250r. PMID 22482496.
- Kuhl, N.; Hopkinson, M. N.; Wencel-Delord, J.; Glorius, F. (2012). 《Beyond Directing Groups: Transition Metal-Catalyzed C H Activation of Simple Arenes》. 《Angew. Chem. Int. Ed.》 51. 10236–10254쪽. doi:10.1002/anie.201203269. PMID 22996679.
- Wencel-Delord, J.; Glorius, F. (2013). 《C–H bond activation enables the rapid construction and late-stage diversification of functional molecules》. 《Nature Chemistry》 5. 369–375쪽. Bibcode:2013NatCh...5..369W. doi:10.1038/nchem.1607. PMID 23609086.
Remove ads
추가 자료
각주
Wikiwand - on
Seamless Wikipedia browsing. On steroids.
Remove ads