Top Qs
Timeline
Chat
Perspective
List of phenyltropanes
From Wikipedia, the free encyclopedia

Remove ads
Phenyltropanes (PTs) are a family of chemical compounds originally derived from structural modification of cocaine. The main feature differentiating phenyltropanes from cocaine is that they lack the ester functionality at the 3-position terminating in the benzene; thus, the phenyl is attached direct to the tropane skeleton (hence the name "phenyl"-tropane) with no further spacer that the cocaine benzoyloxy provided. The original purpose of phenyltropane-related research was to extirpate the cardiotoxicity inherent in the local anesthetic "numbing" capability of cocaine (which stems from the methylated benzoate ester being essential to cocaine's blockage of sodium channels, and which causes topical anesthesia) while retaining stimulant function.[a]
This article may require cleanup to meet Wikipedia's quality standards. The specific problem is: unencyclopediac details in tables: compound-numbers specific to certain references ("7e", for example). (May 2019) |
Phenyltropane compounds present promising avenues of research into therapeutic applications, particularly in regard to addiction treatment. These compounds' uses vary depending on their construction and structure-activity relationship ranging from the treating of cocaine dependency to understanding the dopamine reward system in the human brain to treating Alzheimer's and Parkinson's diseases. (Since 2008 there have been continual additions to the list and enumerations of the plethora of types of chemicals that fall into the category of this substance profile.[2]) Certain phenyltropanes can even be used as a smoking cessation aid (cf. RTI-29). Many of the compounds were first elucidated in published material by the Research Triangle Institute and are thus named with "RTI" serial-numbers (in this case the long form is either RTI-COC-n, for 'cocaine' "analog", or specifically RTI-4229-n of the subsequent numbers given below in this article)[b] Similarly, a number of others are named for Sterling-Winthrop pharmaceuticals ("WIN" serial-numbers) and Wake Forest University ("WF" serial-numbers). The following includes many of the phenyltropane class of drugs that have been made and studied.
 3D rendering of troparil; which comprises a privileged scaffold of among the phenyltropane class of compounds.
3D rendering of troparil; which comprises a privileged scaffold of among the phenyltropane class of compounds. Troparil structure: cf. U.S. patent 5,496,953
Troparil structure: cf. U.S. patent 5,496,953

Remove ads
2-Carboxymethyl esters (phenyl-methylecgonines)
Summarize
Perspective
 Epibatropane[3] containing a nitrogen heteroatom in the benzene ring formation.
Epibatropane[3] containing a nitrogen heteroatom in the benzene ring formation.
 RTI-298
RTI-298 (4′-)para-cis-propenyl-phenyl-methylecgonine. A rare SDRI compound with negligible NET affinity (>2,800.0nM displacement value for NET ligand) that retains significant DAT & SERT (15.0nM & 7.1nM) affinity.
(4′-)para-cis-propenyl-phenyl-methylecgonine. A rare SDRI compound with negligible NET affinity (>2,800.0nM displacement value for NET ligand) that retains significant DAT & SERT (15.0nM & 7.1nM) affinity. C2-C3 unsaturated (non-isomeric, neither α nor β orientated) 2-naphthyl-tropane
C2-C3 unsaturated (non-isomeric, neither α nor β orientated) 2-naphthyl-tropane 1-naphthyl-tropane in its usual (comparably non-standard) boat formation of its tropane ring.
1-naphthyl-tropane in its usual (comparably non-standard) boat formation of its tropane ring.






Like cocaine, phenyltropanes are considered a 'typical' or 'classical' (i.e. "cocaine-like") DAT re-uptake pump ligands in that they stabilize an "open-to-out" conformation on the dopamine transporter; despite the extreme similarity to phenyltropanes, benztropine and others are in suchwise not considered "cocaine-like" and are instead considered atypical inhibitors insofar as they stabilize what is considered a more inward-facing (closed-to-out) conformational state.[5]
Considering the differences between PTs and cocaine: the difference in the length of the benzoyloxy and the phenyl linkage contrasted between cocaine and phenyltropanes makes for a shorter distance between the centroid of the aromatic benzene and the bridge nitrogen of the tropane in the latter PTs. This distance being on a scale of 5.6 Å for phenyltropanes and 7.7 Å for cocaine or analogs with the benzoyloxy intact.[c] The manner in which this sets phenyltropanes into the binding pocket at MAT is postulated as one possible explanation to account for PTs increased behavioral stimulation profile over cocaine.[d]
Blank spacings within tables for omitted data use "no data", "?", "-" or "—" interchangeably.














































|
|
|
(4′-Monosubstituted 2,3-Thiophene phenyl)-tropanes









(3′,4′-Disubstituted phenyl)-tropanes




- ɑas ·HCl (salt)
- bas ·HCl·2 H2O (salt)
- cSingh gives the reverse value with respect to i.e. 1,329 for NET & 320 for 5-HT






-3-(3-ethynylphenyl)-8-methyl-8-azabicyclo(3.2.1)octane-2-carboxylate.svg)




- ɑIC50 determined in Cynomolgous monkey caudate-putamen
- bas ·HCl (salt)
- cas ·HCl·2 H2O (salt)
- dNEN









(2′,4′-Disubstituted phenyl)-tropanes


(3′,4′,5′-Trisubstituted para-methoxyphenyl)-tropanes








ɑN=2
(2′,4′,5′-Trisubstituted phenyl)-tropanes
-3-(4-ethyl-2,5-diiodophenyl)-8-methyl-8-azabicyclo(3.2.1)octane-2-carboxylate.svg)
Remove ads
2-Carbmethoxy modified (replaced/substituted)
Summarize
Perspective
General 2-carbmethoxy modifications
2β-substitutions of p-methoxy-phenyltropanes












ɑN=2
2β-carboxy side-chained (p-chloro/iodo/methyl) phenyltropanes

































- ɑKi value for displacement of [3H]DA uptake.
- bKi value for displacement of [3H]5-HT uptake.
- cKi value for displacement of [3H]NE uptake.
- d[3H]5-HT uptake to [3H]DA uptake ratio.
- e[3H]NE uptake to [3H]DA uptake ratio.
Carboxyaryl



2-Phenyl-3-Phenyltropanes







Carboxyalkyl



Use of a cyclopropyl ester appears to enable better MAT retention than does the choice of isopropyl ester.
Use of a cycBu resulted in greater DAT selectivity than did the cycPr homologue.
2-Alkyl Esters & Ethers
Esters (2-Alkyl)







Ethers (2-Alkyl)






Carboxamides

































✲RTI-183 and RTI-218 suggest possible copy-error, seeing as "CON(OMe)Me" & "CON(Me)OMe" difference between methyl & methoxy render as the same.
Carboxamide linked phenyltropanes dimers





Dimers of phenyltropanes, connected in their dual form using the C2 locant as altered toward a carboxamide structural configuring (in contrast and away from the usual inherent ecgonine carbmethoxy), as per Frank Ivy Carroll's patent inclusive of such chemical compounds, possibly so patented due to being actively delayed pro-drugs in vivo.[3]
Heterocycles
These heterocycles are sometimes referred to as the "bioisosteric equivalent" of the simpler esters from which they are derived. A potential disadvantage of leaving the ββ-ester unreacted is that in addition to being hydrolyzable, it can also epimerize[17] to the energetically more favorable trans configuration. This can happen to cocaine also.

(compound model 34)
Several of the oxadiazoles contain the same number and types of heteroatoms, while their respective binding potencies display 8×-15× difference. A finding that would not be accounted for by their affinity originating from hydrogen bonding.
To explore the possibility of electrostatic interactions, the use of molecular electrostatic potentials (MEP) were employed with model compound 34 (replacing the phenyltropane moiety with a methyl group). Focusing on the vicinity of the atoms @ positions A—C, the minima of electrostatic potential near atom position A (ΔVmin(A)), calculated with semi-empirical (AM1) quantum mechanics computations (superimposing the heterocyclic and phenyl rings to ascertain the least in the way of steric and conformational discrepancies) found a correlation between affinity @ DAT and ΔVmin(A): wherein the values for the latter for 32c = 0, 32g = -4, 32h = -50 & 32i = -63 kcal/mol.
In contrast to this trend, it is understood that an increasingly negative ΔVmin is correlated with an increase of strength in hydrogen bonding, which is the opposing trend for the above; this indicates that the 2β-substituents (at least for the heterocyclic class) are dominated by electrostatic factors for binding in-the-stead of the presumptive hydrogen bonding model for this substituent of the cocaine-like binding ligand.[g]
3-Substituted-isoxazol-5-yl

3-Substituted-1,2,4-oxadiazole
















RTI-4229-470 structure. Highly excited 94 pM DAT signal.[19]
↑ above: 2D skeletal depiction.
↓ below: 3D tube model.
↑ above: 2D skeletal depiction.
↓ below: 3D tube model.






















N.B There are some alternative ways of making the tetrazole ring however; Cf. the sartan drugs synthesis schemes. Bu3SnN3 is a milder choice of reagent than hydrogen azide (cf. Irbesartan).
Acyl (C2-propanoyl)




 Indolyl[21]
Indolyl[21]
cf. the Tamagnan series of phenyltropanes for examples with a methylene unit spacer breaking up the indole.




-8-azabicyclo(3.2.1)octanes.png)



















2β-Acyl-3β-naphthyl substituted




















|
|
Ester reduction
Note: p-fluorophenyl is weaker than the others. RTI-145 is not peroxy, it is a methyl carbonate.

2-Alkane/Alkene






















aKi value for displacement of WIN 35428.
bIC50 value.
Compound 48

para-hydro

para-chloro
Irreversible covalent (cf. ionic) C2 ligands

Irreversible (phenylisothiocyanate) binding ligand (Murthy, V.; Martin, T. J.; Kim, S.; Davies, H. M. L.; Childers, S. R. (2008). "In Vivo Characterization of a Novel Phenylisothiocyanate Tropane Analog at Monoamine Transporters in Rat Brain". Journal of Pharmacology and Experimental Therapeutics. 326 (2): 587–595. doi:10.1124/jpet.108.138842. PMID 18492949. S2CID 5996473.)[23] RTI-76:[24] 4′-isothiocyanatophenyl (1R,2S,3S,5S)-3-(4-chlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate. Also known as: 3β-(p-chlorophenyl)tropan-2β-carboxylic acid p-isothiocyanatophenylmethyl ester.
C2 Acyl, N8 phenylisothiocyanate

HD-205 (Murthy et al., 2007)[25]
Note the contrast to the phenylisothiocyanate covalent binding site locations as compared to the one on p-Isococ, a non-phenyltropane cocaine analogue.
Benztropine based (C2-position hetero-substituted) phenyltropanes





F&B series (Biotin side-chains etc.)
One patent claims a series of compounds with biotin-related sidechains are pesticides.[18]














.svg)
.svg)

.svg)
.svg)






.svg)




Miscellany (i.e. Misc./Miscellaneous) C2-substituents





















Remove ads
C2-truncated/descarboxyl (non-ecgonine w/o 2-position-replacement tropanes)
Summarize
Perspective
Aryl-Tropenes
WO 2004113297, Peters, Dan; Olsen, Gunnar M. & Nielsen, Elsebet Oestergaard et al., "Aza-ring derivatives and their use as monoamine neurotransmitter re-uptake inhibitors", published 2004-12-29, assigned to NeuroSearch AS

Enantioselective nonstandard configurations (non-2β-,3β-)
Summarize
Perspective
β,α Stereochemistry














α,β Stereochemistry


di-chloro; para- & meta- in tandem (α,β configured phenyltropanes)
fumaric acid salts (of α,β configured phenyltropanes)
WO 2004072075, Peters, Dan; Nielsen, Elsebet Oestergaard & Olsen, Gunnar M. et al., "Novel 8-aza-bicyclo[3.2.1]octane derivatives and their use as monoamine neurotransmitter re-uptake inhibitors", published 2004-08-26, assigned to NeuroSearch AS
Remove ads
Arene equivalent alterations
Summarize
Perspective
η6-3β-(transition metal complexed phenyl)tropanes

21b can be prepared from ferrocenes and perrhenate by a double ligand transfer (DLT) reaction.[28]
Unlike metal complexed PTs created with the intention of making useful radioligands, 21a & 21b were produced seeing as their η6-coordinated moiety dramatically altered the electronic character and reactivity of the benzene ring, as well as such a change adding asymmetrical molecular volume to the otherwise planar arene ring unit of the molecule.[1] (cf. the Dewar–Chatt–Duncanson model). In addition the planar dimension of the transition metal stacked arene becomes delocalized (cf. Bloom and Wheeler.[29]).
21a was twice as potent as both cocaine and troparil in displacement of β-CFT, as well as displaying high & low affinity Ki values in the same manner as those two compounds. Whereas its inhibition of DA uptake showed it as comparably equipotent to cocaine & troparil. 21b by contrast had a one hundredfold decrease in high-affinity site binding compared to cocaine and a potency 10× less for inhibiting DA uptake. Attesting these as true examples relating useful effective applications for bioorganometallic chemistry.

The discrepancy in binding for the two benzene metal chelates is assumed to be due to electrostatic differences rather than their respective size difference. The solid cone angles, measured by the steric parameter (i.e. θ) is θ=131° for Cr(CO)3 whereas Cp*Ru was θ=187° or only 30% larger. The tricarbonyl moiety being considered equivalent to the cyclopenta dienyl (Cp) ligand.[1]



- ɑThe binding data fit a two-site model better than a one-site model
- bThe Ki value for the one-site model was 124 ± 10 nM
- cIUPAC: [η6-(2β-carbomethoxy-3β-phenyl)tropane]tricarbonylchromium
- dIUPAC: [η5-(pentamethylcyclopentadienyl)]-[η6-(2β-carbomethoxy-3β-phenyl)tropane]ruthenium-(II) triflate
3-(2-thiophene) and 3-(2-furan)

Thiophenyltropanes

Diaryl

 ZIENT:[32]
ZIENT:[32]


Remove ads
6/7-tropane position substituted
2β-carbomethoxy 6/7 substituted







- ɑIC50 value for displacement of [H3]mazindol. IC50 for cocaine 288 nM for displacement of [H3]mazindol
3-butyl 6/7 substituted














intermediate 6- & 7-position synthesis modified phenyltropanes










Remove ads
8-tropane (bridgehead) position modified
Summarize
Perspective
Nortropanes (N-demethylated)

It is well established that electrostatic potential around the para position tends to improve MAT binding. This is believed to also be the case for the meta position, although it is less studied. N-demethylation dramatically potentiates NET and SERT affinity, but the effects of this on DAT binding are insignificant.[33] Of course, this is not always the case. For an interesting exception to this trend, see the Taxil document. There is ample evidence suggesting that N-demethylation of alkaloids occurs naturally in vivo via a biological enzyme. The fact that hydrolysis of the ester leads to inactive metabolites means that this is still the main mode of deactivation for analogues that have an easily metabolised 2-ester substituent. The attached table provides good illustration of the effect of this chemical transformation on MAT binding affinities. N.B. In the case of both nocaine and pethidine, N-demethyl compounds are more toxic and have a decreased seizure threshold.[34]
ɑThe N-demethylated variant of (i.e. compound code-name after dash)
"Interest in NET selective drugs continues as evidenced by the development of atomoxetine, manifaxine, and reboxetine as new NET selective compounds for treating ADHD and other CNS disorders such as depression" (FIC, et al. 2005).[35]
















ɑThese values determined in Cynomolgus monkey caudate-putamen bThe radioligand used for 5-HTT was [3H]citalopram







Paroxetine homologues
See the N-methyl paroxetine homologues cf. di-aryl phenyltropanes for another SSRI approximated hybrid: the fluoxetine based homologue of the phenyltropane class.







N-replaced (S,O,C)

R-97a (above) & S-97b (below), both examples of interim. synth. prod. in the R/S-90 & 91 series of phenyltropanes; showing the decay of the benzene structure during the synthetic process preceding the creation of like-series of PTs.[1]



-8-methyl-8-azabicyclo(3.2.1)oct-2-ene-2-carbonyl)-3λ⁶-thia-4-azatricyclo(5.2.1.0¹,⁵)decane-3,3-dione.svg)
Mid-synth stage in similar compound preparation as like to above.

The eight position nitrogen has been found to not be an exclusively necessary functional anchor for binding at the MAT for phenyltropanes and related compounds. Sulfurs, oxygens, and even the removal of any heteroatom, leaving only the carbon skeleton of the structure at the bridged position, still show distinct affinity for the monoamine transporter cocaine-target site and continue to form an ionic bond with a measurable degree of reasonable efficacy.
8-oxa bridgehead replacements














8-carba bridgehead replacements



N-alkyl



Bi- and tri-cyclic aza compounds and their uses.[38][39]




































- ɑIC50 for displacement of [3H]cocaine. IC50 for cocaine = 67.8 ± 8.7 (nM)
- bIC50 values for displacement of [3H]WIN 35428
- cIC50 values for displacement of [3H]citalopram
- dThe standard Ki value for the displacement of [3H]GBR 12935, [3H]paroxetine, and [3H]nisoxetine were 27 ± 2, 3 ± 0.2, and 80 ± 28 nM, respectively, for these experiments









Bridged N-constrained phenyltropanes (fused/tethered)
p-methyl aryl front & back N-bridged phenyltropanes


Alternate 2D rendering of compound "42a" (from among the above 'bridged' phenyltropanes) to elucidate the potential overlaying structure of the place inhabited by the constrained nitrogen. Compare JNJ-7925476, tametraline and similar compounds.

RTI-242
- ɑValue for displacement of [3H]WIN 35,428 binding @ DAT
- bValue for displacement of [3H]paroxetine binding to SERT
- cValue for displacement of [3H]nisoxetine from NET
Fused tropane-derivatives as neurotransmitter reuptake inhibitors. Singh notes that all bridged derivatives tested displayed 2.5—104 fold higher DAT affinity than cocaine. The ones 2.8—190 fold more potent at DAT also had increased potency at the other two MAT sites (NET & SERT); NET having 1.6—78× increased activity. (+)-128 additionally exhibited 100× greater potency @ SERT, whereas 132a & 133a had 4–5.2× weaker 5-HTT (i.e. SERT) activity. Front-bridged (e.g. 128 & 129) had a better 5-HT/DA reuptake ratio in favor of SERT, while the back-bridged (e.g. 130–133) preferred placement with DAT interaction.[1] U.S. patent 5,998,405
3,4-Cl2 aryl front-bridged phenyltropanes


- 1-Chloroethyl chloroformate is used to remove N-methyl of trans-aryltropanes.
- 2° amine is reacted with Br(CH2)nCO2Et.
- Base used to abstract proton α- to CO2Et group and complete the tricyclic ring closure step (Dieckmann cyclization).
To make a different type of analog (see Kozikowski patent above)
- Remove N-Me
- Add ɣ-bromo-chloropropane
- Allow for cyclization with K2CO3 base and KI cat.
C2 + C3 (side-chain) fused (carboxylate & benzene conjoined)
-3-(4-methylphenyl)-9,18-diazapentacyclo(9.7.0.0²,⁸.0⁵,⁹.0¹²,¹⁷)octadeca-1(11),12(17),13,15-tetraene.svg)
-15-methyl-15-azatetracyclo(10.2.1.0²,¹⁰.0⁴,⁹)pentadeca-4(9),5,7-trien-3-one.svg)
(1R,2S,10R,12S)-15-methyl-15-azatetracyclo(10.2.1.02,10.04,9)pentadeca-4(9),5,7-trien-3-one[3]
C3 to 1′ + 2′ (ortho) tropane locant dual arene bridged

Parent compound of a series of spirocyclic cocaine benzoyl linkage modification analogs created by Suzuki coupling method of ortho-substituted arylboronic acids and an enol-triflate derived from cocaine; which technically has the three methylene length of cocaine analogues as well as the single length which defines the phenyltropane series. Note that the carbomethoxyl group is (due to constraints in synthetic processes used in the creation of this compound) alpha configured; which is not the usual, most prevalent, conformation favored for the PT cocaine-receptor binding pocket of most such sub-type of chemicals. The above and below depictions show attested compounds synthesized, additionally with variations upon the Endo–exo isomerism of their structures.[40]

Remove ads
Cycloalkane-ring alterations of the tropane ring system
Summarize
Perspective
Azanonane (outer ring extended)
3-Phenyl-9-azabicyclo[3.3.1]nonane derivatives
To better elucidate the binding requirements at MAT, the methylene unit on the tropane was extended by one to create the azanonane analogs.[i] Which are the beginning of classes of modifications that start to become effected by the concerns & influences of macrocyclic stereocontrol.
Despite the loosened flexibility of the ring system, nitrogen constrained variants (such as were created to make the bridged class of phenyltropanes) which might better fit the rigid placement necessary to suit the spatial requirements needed in the binding pocket were not synthesized. Though front-bridged types were synthesized for the piperidine homologues: the trend of equal values for either isomers of that type followed the opposing trend of a smaller and lessened plasticity of the molecule to contend with a rationale for further constraining the pharmacophore within that scope. Instead such findings lend credence to the potential for the efficacy of fusing the nitrogen on an enlarged tropane, as like upon the compounds given below.






Azabornane (outer ring contracted)
3-Phenyl-7-azabicyclo[2.2.1]heptane derivatives
Ring-contracted analogs of phenyltropanes did not permit sufficient penetration of the phenyl into the target binding site on MAT for an affinity in the efficacious range. The distance from the nitrogen to the phenyl centroid for 155a was 4.2 and 155c was 5.0 Å, respectively. (Whereas troparil was 5.6 & compound 20a 5.5 angstroms). However piperidine homologues (discussed below) had comparable potencies.[j]
heptane.png)
The non-carboxylic (and DAT substrate, releasing agent) variant of exo-2-phenyl-7-azabicyclo(2.2.1)heptane-1-carboxylic acid (N.B. the carboxy in the latter shares the C1 tropane position with the two carbon nitrogen containing bridge; sharing in the leftmost (R) substitution of the above depiction & unlike the placement on the tropane for either the carbmethoxy or phenyl ring of the azabornane analogues given in this section)
With the carboxy ester function removed the resultant derived compound acts as a DAT substrate drug, thus an amphetaminergic releaser of MAT & VMAT, yet similar to phenyltropanes (that usually are only re-uptake ligands)[41] cf. EXP-561 & BTQ.
Azabornanes with longer substitutions at the 3β-position (benzoyloxys alkylphenyls, carbamoyls etc.) or with the nitrogen in the position it would be on the piperidine homologues (i.e. arrangements of differing locations for the nitrogens being either distal or proximal within the terms required to facilitate the framework of the compound to a correlative proportion, functional for the given moiety), were not synthesized, despite conclusions that the nitrogen to phenyl length was the issue at variance enough to be the interfering factor for the proper binding of the compressed topology of the azabornane. Carroll, however, has listed benzoyloxy azabornanes in patents.[3]





Piperidine homologues (inner two-carbon bridge excised)
Piperidine homologues had comparable affinity & potency spreads to their respective phenyltropane analogues. Without as much of a discrepancy between the differing isomers of the piperidine class with respect to affinity and binding values as had in the phenyltropanes.
p-chloro & related (piperidine homologues of phenyltropanes)









Heterocyclic N-Desmethyl[43]
-3-(3-methyl-(1,2,4)oxadiazol-5-yl)-piperidine.png)
Naphthyl & related (piperidine homologues of phenyltropanes)













distal-nitrogen 'dimethylamine' (piperidine-like cyclohexyl homologues of phenyltropanes)
Source:[3]



cf. Fencamfamine
Remove ads
Radiolabeled
Summarize
Perspective


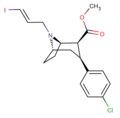 IPT (N-3-iodoprop-(2E)-ene-2β-carbomethoxy-3β-(4′-chlorophenyl)tropane), can be radiolabeled with 123I or 125I and used as a ligand to map several MATs
IPT (N-3-iodoprop-(2E)-ene-2β-carbomethoxy-3β-(4′-chlorophenyl)tropane), can be radiolabeled with 123I or 125I and used as a ligand to map several MATs
 JHC1-64.[53] A fluorescent analog, similar in its long chain off of the nitrogen bridge similar to the transition metal phenyltropane types.
JHC1-64.[53] A fluorescent analog, similar in its long chain off of the nitrogen bridge similar to the transition metal phenyltropane types.
-ene-2β-carbomethoxy-3β-(4′-chlorophenyl)tropane.png)


Transition metal complexes
These compounds include transition metals in their heteroatomic conformation, unlike non-radiolabel intended chelates where their element is chosen for intrinsic affectation to binding and function, these are tagged on by a "tail" (or similar) with a sufficient spacer to remain separated from known binding properties and instead are meant to add radioactivity enough to be easily tracked via observation methods that utilize radioactivity. As for anomalies of binding within the spectrum of the under-written kinds just mentioned: other factors not otherwise considered to account for its relatively lower potency, "compound 89c" is posited to protrude forward at the aryl place on its moiety toward the MAT ligand acceptor site in a manner detrimental to its efficacy. That is considered due to the steric bulk of the eight-position "tail" chelate substituted constituent, overreaching the means by which it was intended to be isolated from binding factors upon a tail, and ultimately nonetheless, interfering with its ability to bind. However, to broach this discrepancy, decreasing of the nitrogen tether at the eight position by a single methylene unit (89d) was shown to bring the potency of the analogous compound to the expected, substantially higher, potency: The N-methyl analog of 89c having an IC50 of 1.09 ± 0.02 @ DAT & 2.47 ± 0.14 nM @ SERT; making 89c upwards of thirty-three times weaker at those MAT uptake sites.[k]






- ɑIUPAC: [2-[[2-[[[3-(4-chlorophenyl)-7-methyl-8-azabicyclo[3,2,1]oct-2-yl]methyl]-(2-mercaptoethyl)amino]ethyl]amino]ethanethiolato-(3—)-N2, N2′, S2, S2′]oxo-[1''R''-(''exo'', ''exo'')]-[99mTc]technetium
- bR- & S- isomer values are Ki (nM) for displacement of [125I]IPT with technetium-99m replaced by rhenium
- cIC50 (nM) values for displacement of [3H]WIN 35428 with ligand tricarbonyltechnetium replaced with rhenium. (IC50 for WIN 35428 were 2.62 ± 1.06 @ high affinity binding & 139 ± 72 @ low affinity binding sites)
- dKi value for displacement of [125I]IPT radioligand.
Remove ads
Select annotations of above
Phenyltropanes can be grouped by "N substitution" "Stereochemistry" "2-substitution" & by the nature of the 3-phenyl group substituent X.
Often this has dramatic effects on selectivity, potency, and duration, also toxicity, since phenyltropanes are highly versatile. For more examples of interesting phenyltropanes, see some of the more recent patents, e.g. U.S. patent 6,329,520, U.S. patent 7,011,813, U.S. patent 6,531,483, and U.S. patent 7,291,737.
Potency in vitro should not be confused with the actual dosage, as pharmacokinetic factors can have a dramatic influence on what proportion of an administered dose actually gets to the target binding sites in the brain, and so a drug that is very potent at binding to the target may nevertheless have only moderate potency in vivo. For example, RTI-336 requires a higher dosage than cocaine. Accordingly, the active dosage of RTI-386 is exceedingly poor despite the relatively high ex vivo DAT binding affinity.
Remove ads
Sister substances
Many molecular drug structures have exceedingly similar pharmarcology to phenyltropanes, yet by certain technicalities do not fit the phenyltropane moniker. These are namely classes of dopaminergic cocaine analogues that are in the piperidine class (a category that includes methylphenidate) or benztropine class (such as Difluoropine: which is extremely close to fitting the criteria of being a phenyltropane.) Whereas other potent DRIs are far removed from being in the phenyltropane structural family, such as Benocyclidine or Vanoxerine.
Most any variant with a tropane locant—3-β (or α) connecting linkage differing from, e.g. longer than, a single methylene unit (i.e. "phenyl"), including alkylphenyls (see the styrene analog, first image given in example below) is more correctly a "cocaine analogue" proper, and not a phenyltropane. Especially if this linkage imparts a sodium channel blocker functionality to the molecule.
Remove ads
See also
References
External links
Wikiwand - on
Seamless Wikipedia browsing. On steroids.
Remove ads